計算化学とは?基礎入門・手法と応用(新薬・材料設計)
計算化学の基礎から手法、実務的応用まで詳解。新薬・材料設計に役立つ理論と実例をわかりやすく解説。
計算化学は、化学の一分野であり、化学的な問題を解決するためにコンピュータ科学を使用します。これらのプログラムは、分子や固体の構造や性質を計算します。計算化学は通常、化学実験で得られた情報を補完するものです。これは、まだ観測されていない化学現象を予測することができます。新薬や材料の設計に広く利用されています。
計算化学は、構造(分子の原子の予想される位置)、絶対・相対(相互作用)エネルギー、電子電荷分布、双極子や高次多極子モーメント、振動周波数、反応性や他の分光学的量、他の粒子との衝突のための断面積などを予測することができます。
計算化学は、静的なシステムと動的なシステムの両方を対象としています。いずれの場合も、研究対象となるシステムのサイズが大きくなると、コンピュータの時間やその他のリソース(メモリやディスクスペースなど)の使用量も大きくなります。このシステムは、単一の分子、分子群、固体のいずれであっても構いません。計算化学の手法には、高精度なものから非常に近似的なものまで様々なものがあります。高精度な方法は、通常、小さなシステムでのみ実現可能です。
画像ギャラリー
8 画像


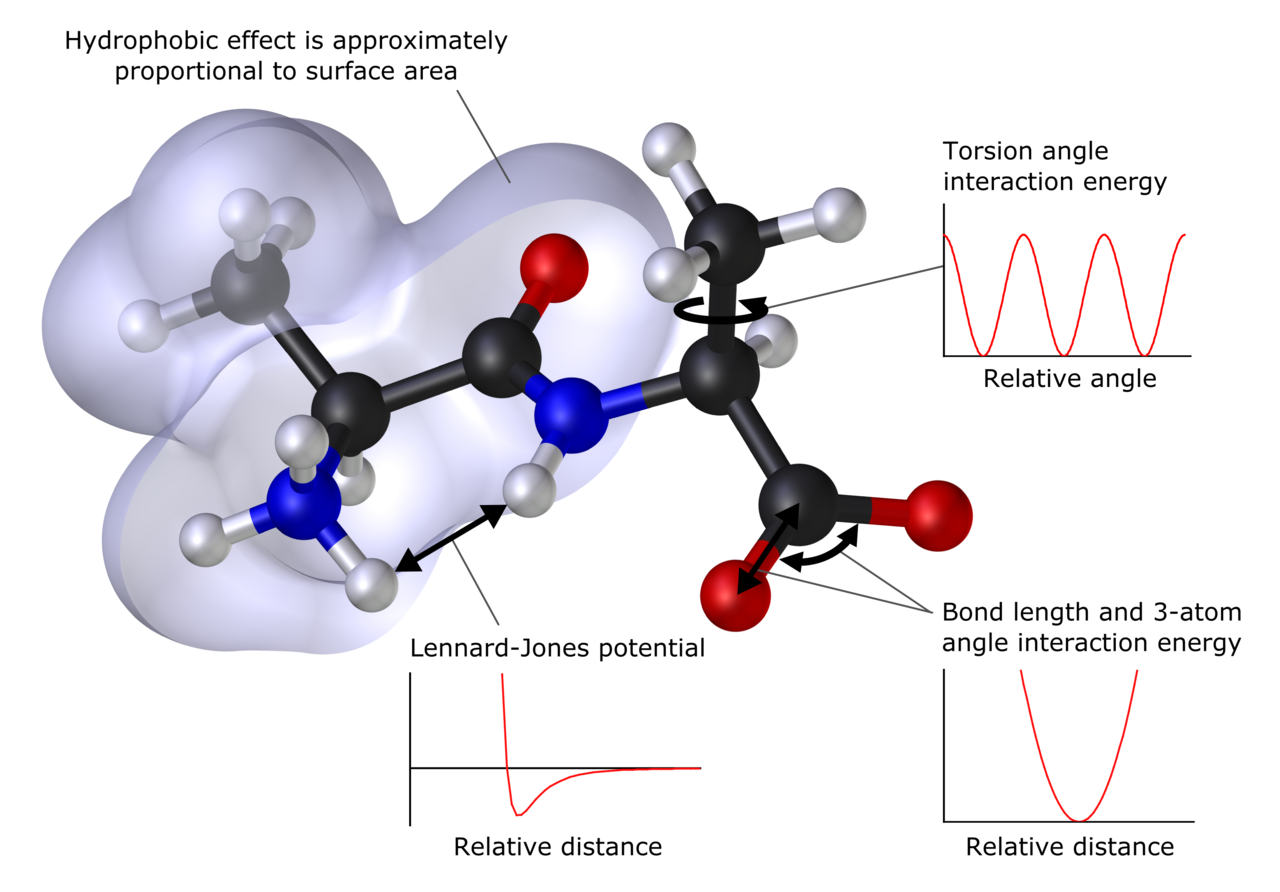


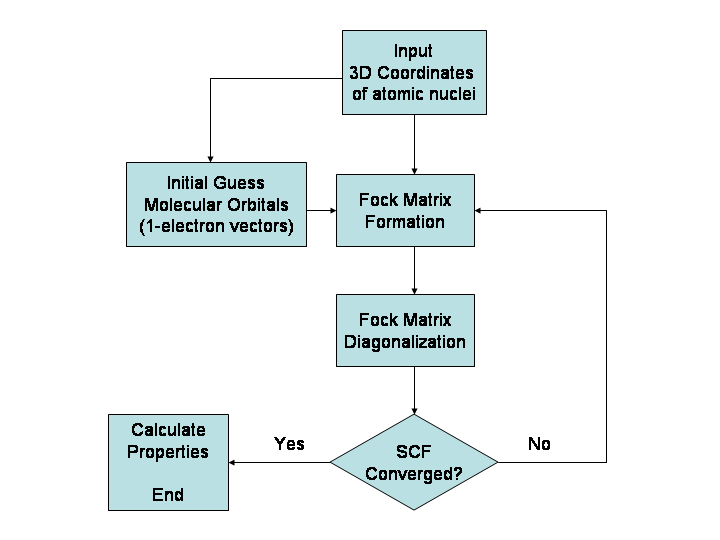
主な計算手法(概要)
- 量子化学(ab initio)法:波動関数に基づく厳密性の高い方法。ハートリー–フォック(HF)や、それに続く相関を扱うMP2、CCSD(T)などがあります。精度は高いが計算コスト(スケーリング)が大きい。
- 密度汎関数法(DFT):電子密度を主変数とする手法で、実用的な精度と計算効率の良さのため広く用いられます。交換・相関汎関数(B3LYP、PBE、ωB97X など)の選択が結果に大きく影響します。
- 半経験的手法:理論式に実験データやパラメータを導入して近似する方法。大型系を素早く扱えますが、系依存性があるため注意が必要です。
- 分子力学(MM)/力場:原子を点とし、結合、角度、絡み合いなどを古典力学で表現します。大規模分子や長時間の動的挙動(生体分子、ポリマー、材料)に適します。代表的な力場にAMBER、CHARMM、OPLSなどがあります。
- 分子動力学(MD):力場を用いて原子運動を時間発展させ、温度・圧力下でのダイナミクスや統計的性質を調べます。拡張手法として加速サンプリング法やフリーエネルギー計算法(FEP、TI、メトロポリス法)があります。
- QM/MM ハイブリッド:反応中心など重要部分を量子化学で、残りを力場で扱う手法。触媒や酵素反応のシミュレーションで有用です。
- モンテカルロ法:確率的サンプリングにより平衡性状を評価する手法。MDよりも効率よく構造空間を探索できる場合があります。
計算で予測できる主な量
- 分子・結晶構造の最安定配置、結合長や角度
- エネルギー(単一点エネルギー、相対エネルギー、反応座標上の遷移状態エネルギー)
- 電子密度や電荷分布、双極子・多極子モーメント
- 振動周波数(IRスペクトルの予測)、NMR化学シフト、UV/Visスペクトルなどの分光学的性質(分光学的量)
- 反応性や活性化エネルギー、触媒サイクルの解析(遷移状態の同定)
- 衝突断面積、拡散係数、輸送物性などの動的・輸送現象
応用例:新薬・材料設計を中心に
- 新薬探索:リガンドの結合モード予測(ドッキング)、結合自由エネルギーの定量的評価(FEP/TI)、反応機構解析、代謝予測、薬物候補の物性予測(溶解度、pKaなど)。計算は候補のスクリーニングや実験計画の最適化に有用です。
- 材料設計:固体のバンド構造、欠陥エネルギー、相安定性、触媒表面の反応性、電池材料のイオン拡散など、原子・電子レベルでの設計指針を提供します。
- 触媒開発:反応経路の可視化、活性部位の同定、遷移状態エネルギープロファイルから触媒設計を導く。
- 分光学的解析:実験スペクトルのピーク割り当てや構造同定の補助。
計算資源とスケーリング
計算量は手法によって大きく異なります。たとえば、HFや一部のDFTは比較的計算負荷が低い一方で、ポストHF法(CCSD(T)など)は計算量が原子数Nに対してN^6〜N^7と急増します。力場ベースのMDは一原子当たりのコストは低いですが、長時間スケールのサンプリングが必要なため総コストは高くなり得ます。
実務的ポイント:収束(基底関数の大きさやカットオフエネルギー)、サンプリング時間、温度・圧力条件などの設定が結果に影響します。計算コスト節約のために多段階戦略(粗い手法で絞り込み、精密手法で最終評価)を使うのが一般的です。
注意点・限界
- どの手法にも近似が含まれるため、常に誤差や系依存性が存在します。特にDFTでは汎関数の選択によるバラツキが問題になります。
- 基底関数や力場パラメータ、境界条件(周期境界など)による誤差(例:基底関数重複誤差=BSSE)が生じ得ます。
- 動的・統計的性質を評価するには十分なサンプリング(長時間MDや複数の独立試行)が必要で、計算コストと時間がかかります。
- 実験との比較・検証が不可欠。計算はあくまで仮説・設計支援であり、実験データでの確認が必要です。
実際のワークフローとツール
典型的な流れ:初期構造取得 → ジオメトリ最適化 → 周波数解析(安定性確認、熱力学量)→ 必要に応じて遷移状態探索や自由エネルギー計算 → 結果解析・実験比較。ソフトウェア例としては、Gaussian、ORCA、NWChem(量子化学)、VASP、Quantum ESPRESSO(固体・第一原理)、GROMACS、AMBER、LAMMPS(分子力学/MD)などがあります。
今後の動向
機械学習を用いたポテンシャルや物性予測、GPU による高速化、高スループット自動設計ワークフロー、量子コンピュータの適用可能性など、計算化学は計算能力の向上とともに急速に発展しています。これにより、より大きな系や長時間スケールの現象の取り扱いが容易になり、実験との連携も一層進むと期待されています。
まとめ(実務的アドバイス)
- 目的に応じて手法を選ぶ:精度が必要なら高レベル量子化学、広い空間と長時間が必要なら力場+MD。
- 妥当性検証:基底関数や汎関数、力場の妥当性を小スケールでテストしてから本計算へ進める。
- 結果は相対値・傾向として解釈:絶対値よりも相対比較(候補AとBの差)で有用なことが多い。
- 実験との協働:計算で得た仮説は実験で検証し、モデルの改良につなげる。

関連ページ
- バイオインフォマティクス
- 統計力学
質問と回答
Q:計算化学とは何ですか?
A: 計算化学は、化学的な問題を解決するためにコンピュータサイエンスを使用する化学の一分野です。分子や固体の構造や性質を計算し、まだ観測されていない化学現象を予測したり、新しい薬や材料を設計したりするのに利用されます。
Q:計算化学では、どのような系を見ているのですか?
A: 計算化学は、静的な系と動的な系の両方を対象としています。システムは、単一の分子、分子のグループ、または固体のいずれでもかまいません。
Q:計算化学はどのような情報を提供するのですか?
A: 計算化学は、構造(原子の位置)、絶対および相対エネルギー、電子電荷分布、双極子および高次多極子モーメント、振動数、反応性またはその他の分光学的量、および他の粒子との衝突の断面積といった情報を提供することができます。
Q:計算化学で使われる手法の精度はどのくらいですか?
A: 計算化学で使用される手法の精度は、高精度なものから非常に近似的なものまで様々です。高精度の手法は、一般的に小さな系でのみ実現可能です。
Q: 計算化学は、実験データをどのように補完するのですか?
A: 通常、計算化学は化学実験によって得られた情報を補完するものです。まだ実験的に観測されていない結果を予測するために使用することができます。
Q: 研究対象のシステムの大きさは、必要なコンピュータ時間に影響しますか?
A: はい。研究対象のシステムのサイズが大きくなればなるほど、解析に必要なコンピュータの時間や、保存に必要なメモリやディスクスペースなどの資源も大きくなります。
関連項目
著者
AlegsaOnline.com 計算化学とは?基礎入門・手法と応用(新薬・材料設計) Leandro Alegsa
URL: https://ja.alegsaonline.com/art/22297